Diseases and Conditions › Heart Diseases › New reason for and the mechanisms of cardiac electrical instability. New defibrillation mechanism.
New reason for and the mechanisms of cardiac electrical instability.
New defibrillation mechanism.
Rustam G. Habchabov
Dagestan state medical academy, Russia
1, st. Lenina, Makhachkala city, 367000
PhD (medical)
E-mail: rustam033@gmail.com
Abstract. The reason for development of life-threatening arrhythmias leading to sudden and total death provoked by cardiac diseases is still unclear, even though cardiologic research in this regard is being carried all over the word. A damaging of the connective insulation cover of the conductive heart path accompanied by ectopic nodes oxidation may result in life-threatening arrhythmias. Such reason for it as the cardiac electrical instability hasn’t been examined by anyone before. The connective insulation cover prevents a defibrillating electric discharge from penetrating the myocardium. Simultaneously, nobody took the conductive nervous system into account.
Keywords: electric instability of heart; trembling; fibrillation; defibrillation; nervous system; syndrome WPW-CLC.
Cardiovascular diseases (CVDs) remain main causes of invalidity and death of working-age population all over the world. In Russia CVDs frequency is increasing, while our country is considered to be one of the world leaders in death caused by CVDs. All this makes us search for modern and efficient methods of diagnostics, treatment and prevention of the cardiac electrical instability (CEI).
At a young age patients with rheumatic disease, myocarditis, mitral valvular disease demonstrate flutter and fibrillation, while geriatric and older patients demonstrate it more often if they are diagnosed with ischemic heart disease (IHD), myocardial infraction (MI), chronic cardiac failure, mitral stenosis, hypertensive disease, cardiomyopathy, myocarditis, etc. [4].
The condition specifying lethal arrhythmias is structural cardiac pathology (SCP) turning into an instable substrate as affected by various functional factors. Such structural changes conditioning the development of life-threatening arrhythmias (LTA) may be as follows: express hypertrophy, dilatation, cardiac aneurysm, myocardial necrosis and sclerosis, inflammation accompanied by myocardial tissue edema, etc. According to the data of many researchers, these changes constitute the anatomical substrate with various LTA development mechanisms [9].
Many authors that conducted CEI research often concentrate only on myocardial necrotizing and cicatricial lesions (NCL) in IHD patients. Yet such authors ignore other patients with other CVDs that are exposed to total and sudden cardiac death (SCD), too, for no apparent reason.
It should be mentioned that practically all arrhythmias are easy to be analyzed, except for the reason and the mechanism for flutter and fibrillation development. Moreover, the definition of atrial fibrillation is misleading because of the contradictions contained in electrocardiography (ECG) textbooks.
According to the definition contained in ECG textbooks [4,5], atrial and ventricular flutter is regular rhythmical movement of a potent impulse following one and the same path with simultaneous re-entry mechanism development (the reason and the method of such macro-re-entry mechanism being unclear to the researchers at that moment). Atrial and ventricular fibrillation is irregular excitation and contraction of some groups of muscle fibers, each of them being a sort of ectopic impulse site (such definition in ECG textbooks does not seem to be too correct for students and young scientists). Having read the definition and returning back to the “automatism function†chapter one may read that the automatism function is pertinent to Sinoatrial Node cells (SAN) and the conductive system of heart: atrioventricular AV connection, the connective system of atriums and ventricles, the contractive myocardium being deprived of the automatism function [4,5]. All this testifies to the fact that in the event of atrial fibrillation “some groups of muscle fibers†cannot be generated, even if the myocardium or cardiomyocytes change their properties. The “electric myocardial instability†is non-existent because the myocardium can only conduct electric impulses. Some may object that even though the myocardium is deprived of electrical activity, yet it may have a nonhomogeneity area associated with NCL that may prevent it from conducting electricity. Virtually, the electrical impulses avoid the area easily, even in the event of extensive MI, even if there are no serious problems of the cardiac conduction path (CCP). Atrial fibrillation itself is conditioned by multiple generations of ectopic focuses (EF) of a lesser power located in distal CCP and with the same number of colliding micro-re-entry waves. In fact, the myocardium cannot generate it and the ectopic focuses are not chaotically scattered along the myocardium, as some might believe. All EFs are placed along the CCP, as a string of beads, and have the same connective insulation cover (CIC) starting from the SAN. If this cover is intact, electric impulses cannot leave the myocardium. It’s only Purkinje fibers that lack such cover and it’s due to them that the myocardium is excitated.
First of all one should mention that CCPs are placed in the same manner as electric wires in walls of our houses are. They also have an insulation cover. Yet the wires located in walls are immovable and they almost do not wear out while a working myocardium with SCP causes them to wear out soon resulting in a block and/or damaging of CIC only.
In some cases of express SCP over-distension breaking or tearing of a CIC of one of the main proximal CCP may occur. It activates the nearest potent EF with flutter development. This causes a potent electrical impulse to reach the myocardium along the path of least resistance through such damaging and trigger the macro re-entry mechanism. This is how large F flutter waves are formed (besides, the same mechanism is described for Alzheimer disease when atherosclerosis damages the nerve’s medullary sheath and nerve impulses diffuse into the surrounding tissue. Is CCP’s atherosclerotic damaging also possible?).
One may wonder if the proposed mechanism of flutter development is correct. The fact is that the most potent ectopy source in supraventricular paroxysmal tachycardia and atrial flutter is located in the atrium (atrial fibrillation cannot be accounted for; it is conditioned by multiple ectopy). Yet ECG demonstrates a different picture. Why? It may be explained by the fact that in supraventricular paroxysmal tachycardia an electric impulse has to overcome many obstacles passing through a number of small conductive paths and ectopic nodes within the atriums and it reaches the myocardium as a not-so-potent impulse to spin a macro-re-entry wave. In atrial flutter a potent electric impulse reaches the myocardium prematurely, through a CIC damaging, encountering no obstacles and spins a macro re-entry wave. It’s most probable that the lesser part of electric impulse that passes along the conductive paths and exits Purkinje fibres prevents the greater part of electric impulse that reaches the myocardium prematurely, through a CIC damaging, from entering the conductive paths of atriums, encountering a counter-propagating depolarization wave there. All this forms a macro re-entry wave. Such atrial pre-excitation with the macro re-entry mechanism results in a small difference in the cardiac rate between the supraventricular paroxysmal tachycardia and atrial fibrillation. In other words, most frequently paroxysmal tachycardia damages CIC CCP (the conductive paths of Venkenbach, Bakhman and Torel, and the Bundle of His and branches in the ventricles) and becomes flutter. Yet the ectopy source remains to be the same, so it’s the same type of arrhythmia having different mechanisms of action. For example, I have mentioned the atrial process which is exactly the same as the ventricular process.
What is the cause for potent EF activation with flutter development in the event of CIC damaging? If CICs are intact, CCPs are not subject to oxidation. Purkinje, the famous researcher, described transitional T-cells located between the conductive B-cells (Purkinje cells) and the myocardium. He thought that their main function was to conduct electric impulses but their main function is more likely to consist in forming an antioxidative barrier for the conductive B-cells. Myocardial intercellular fluid shall not get into CCP, so the two conductors having a completely different structure have to have an “adaptor†among them that would prevent them from oxidation. If we eliminate the T-cells, the conductivity of electric impulses between B-cells and the myocardium shall remain the same for some time but Purkinje fibres would be exposed to oxidation. With the course of time oxidation would produce irrevocable consequences. Nature did not provide for transitional T-cells for emergency cases. It results in rapid oxidation of the nearest proximal EF with concomitant flutter development. Further the oxidation processes shall reach other distal EF and flutter will gradually turn into fibrillation.
In many cases flutter does not precede atrial fibrillation. It happens because in fibrillation CIC of mid-distal CCPs are to be damaged most frequently (atrial or ventricular Purkinje fibres and braches), especially in NCL. Multiple T-cells damaging is also possible and results in the oxidation of more than one less potent EFs and micro-re-entry mechanism’s development. Thus one may say that the reason for CEI development is the same for flutter and fibrillation.
Any sudden myocardial movements or lesions may trigger CIC CCP damaging in SCP: all types of tachycardia, extrasystoles, blocks, abrupt blood pressure (BP) increase, myocardial contractility increase (especially if accompanied with pathologically decreased contractility), NCL, etc.
As for the focalization, the most thin and easy-to-be-damaged ventricular segments are the right bundle and the left anterior bundle branch. Then main stem of the left branch, the Bundle of His and, finally, the right anterior bundle branch may be named in the descending order. Yet conductivity may be damaged in any segment or in a few segments simultaneously [5].
Other main factor in arrhythmia development is associated with the affected contractility and excitation, most frequently in SCP patients:
1. Organic or functional weakness of the ectopic node (I, II, III block – by AV block type) that result in weak electric impulses arriving to the distal node and its eventual activation.
2. Over-distension of the conductive path (especially in places where ectopic nodes are located) in express hypertrophies, dilatations, etc. lead to the extension of conductivity which also enhances a delay in electric impulses and activates EF.
3. Myocardial inflammation slows down the conductivity and results in ectopic activity development.
The example may be as follows: a city has been growing for 20-30 years because its suburbs (myocardial hypertrophy, dilatation) were growing, too. If the public utility network is lacking in the new districts, the inhabitants of such districts would complain of the lack of electricity. The same is true for the heart. The central nervous system tries to increase the cardiac rate and the potency of electric impulses to compensate for it. Sometimes it is enough. If not, ectopic arrhythmia tries to compensate for such insufficiency.
In some cases electric cardioversion may stop atrial fluttering and fibrillation at their early stages (paroxysmal, persistent). Powerful SAM stimulation suppressing other generation centers takes place. For some time the oxidative influence on EF becomes not important and minute CIC damaging is regenerated after a while. High doses of antyarrhytmic medicines at early stages of flutter and fibrillation are also capable of suppressing ectopic activity with gradual regeneration of minute CIC damaging and sinus rhythm restoration.
Preventive use of repairers in SCP patients may prevent CIC CCP damaging, such repairers being: potassium orotate, ATP and solcoseryl. It goes without saying that basic treatment of patients with atrial flutter and fibrillation shall remain at the previous level– cardioversion and antyarrhytmic drugs. Though additional alkalizing medicines and repairers, with the exception of aspirin and other acid-containing substances, for the period of flutter and fibrillation treatment may contribute into restoring cardiac electrical stability.
To continue CIC and the conductive system’s issue, let me add a few speculations of my own. Electric defibrillation discharges are considered to influence the heart directly, but it’s an error! In the course of defibrillation human anatomy prevents electric discharges from entering the myocardium and exiting from it because of the external epicardial and endocavitary endocardial cardiac layer that has CIC. Electric discharges are most likely to affect the heart in an indirect manner, through multiple nervous receptors. Electric impulses strive to reach the CNS and then brain activation concerning all its aspects, including the sympathic nervous system (SNS) of the heart, starting from and SAN and b-adrenoreceptors stimulation with catecholamines (adrenaline, noradrenaline) discharge. Nobody thought that external electric currents can also take nervous paths in the course of defibrillation.
Now I’m going to present a theory which would seem incredible to many scientists but it has a right to exist. It’s still unknown where and how electric impulses are formed in the nervous system. Electric impulses seem more likely not to be generated by the nervous system but to be sent to it from all the nervous receptors, starting from the SAN and ending up with ventricular myocardium where the electric impulse comes to a dead end. Further electricity is conducted by the conductive (afferent) nerve fiber to various CNS divisions. One should understand that ECG and electroencephalogram (EEG) recording have nothing in common because they record excitation in different structures of the organism. Such circular relationship of the heart and the nervous system is more reasonable that two separate electric systems which would have inevitably come in conflict and lead to a short circuit in one and the same organism.
One may wonder why the nervous system needs electricity. Only for instant processing of the received information and for sending it back. Passive work of the nervous system may be performed without electricity, as it happens during heart transplantation when a patient remains on artificial blood circulation for a few hours. EEG records passive work of the brain which should give an impulse for the transplanted heart to start. In some cases it’s not enough and defibrillation is implemented.
Further we are going to analyze literature sources and main views of the authors and researchers with reference to the CEI issue. First of all one should mention that many authors write “electrical instability of myocardium†while in reality it’s more correct to write “cardiac electrical instabilityâ€.
Life-threatening LTA are caused by a combination of reasons predisposing to electrical instability of myocardium: a substrate (structural cardiac disease) modulating the dysfunction of the autonomous nervous system and LTA triggering factors. The morphologic substrate creating post-MI nonhomogeneity of impulse conductivity is a myocardial area adjoining the necrotized tissue formed by intertangled spots of healthy myocardial fibers and the connective tissue. In this place the impulse connective path is prolonged because the spots of connective tissue serve as barriers to the excitation wave and the conductivity is slowed down due to the affected parallel orientation of muscle fibers. Thus myocardial areas with delayed ventricular depolarization may be anatomic and physiological substrate for the re-entry – main mechanism of LTA development [1,3]. The author of this study was close to understand the real situation but the source of LTA development is not the borderline myocardium but CIC CCP necrotic damaging in the area with concomitant development of flutter and/or atrial fibrillation.
Following the results of the work of J.D. Kramer et al., long impulse spin path is not necessary a small diameter of myocardial tissue with its electrophysiological properties altered by acute myocardial ischemia or with a heterogeneous structure resulting from fibrous and necrotizing changes is enough to trigger the re-entry mechanism [11]. Fibers of the conductive system with EF are more hypoxia-resistant and myocardial ischemia does not trigger the re-entry mechanism in them, while fibrous-necrotizing changes are capable of damaging CIC CCP and triggering arrhythmia with the re-entry mechanism.
As a rule, in the course of the conducted study sudden ventricular tachycardia (VT) or ventricular fibrillation (VF) with the maintained ejection fraction was observed in patients with implanted cardioverters defibrillators; if the contractility is decreased before VT or VF attacks, normally gradual increase of ventricular ectopic activity is to be noted [7]. It goes without saying that cardiac failure is an essential arrhythmogenic factor and a risk marker of sudden arrhythmic death in IHD patients [12]. A Cardiac aneurysm, post-infarction cicatricial changes and clinical manifestations of cardiac failure make the adverse outcome more probable. Left ventricular contractility decrease increases SCD risk not only in IHD but also in patients with other cardiac diseases [8, 9]. Ejection fraction less than 40%, nonsustained ventricular tachycardia (VT) diagnosed by Holter monitoring and electrophysiological study in patients that had an acute MI in their medical history are remaining to be the main predictive markers of high SCD risk [10]. Such combination of two SCD risk factors as frequent ventricular arrhythmia and left ventricular dysfunction with an ejection fraction decrease <40% is especially unfavorable. According to the data of GISSI-2 research, a risk of sudden arrhythmic death in this case increases 16-fold [2, 15].
Besides that factors that were mentioned above, other sudden death risk factors are known, autonomic imbalance of the heart with prevailing sympathetic activity, in particular. The most important markers of the state is a decrease of the cardiac rate variability (CRV) and also such factors as a continued prolongation of the QT interval dispersion and late ventricular potentials (LVP) [13,14].
I can only add a few summarizing conclusion from the PhD thesis I defended with reference to CRV, QT interval dispersion and LVP. Initial degradation of CRV parameters in patients during the post-infarction period is associated with their anxious and depressive state after acute MI. Depression is diagnosed in 82% of patients in the post-infarction period [6]. Such patients are afraid of death and are anxious for their health, they do not perceive their environment with joy. On the contrary, they become reserved and their SNS becomes more active than the parasympathetic nervous system (PNS). As positive psycho-emotional state of the patients is activated, they start to overcome depression and PNS functioning becomes stable, too. This results in the sinus rhythm variations. One should note that a positive psycho-emotional state of a post-MI patient may become negative and even stress condition. This, in turn, leads to SNS hyperarousal. Combined with atherosclerotic changes in coronary arteries, SNS hyperarousal may result in a spasm of coronary arteries and appearance of new necrotic lesions in the myocardium. This is why SAN nervous regulation is more perfect due to the simultaneous operations of the sympathetic and the parasympathetic divisions of the autonomous nervous system.
LVP in the post-infarction period is improved less significantly than the QT interval dispersion parameters. This may be conditioned by the fact that LVP is more associated with the CCP block (His bundle branches and/or main stems of Purkinje fibres) that are completely blocked by the nonhomogeneous necrotized area and further cicatricial changes of the myocardium which make the electric impulse return and reach the myocardium through other CCPs.
As different from LVP, the regeneration processes of the QT interval are to a lesser extent associated with CCP pathological changes. In the acute MI, the QT interval dispersion is predominantly slowed down by necrosis and a reinfarction myocardial area having a nonhomogenious area which is 20-40% larger than in the cicatricial period. Further it improves re-polarization processes. In single cases of LVP improvement in the post-infarction period rare cases of the organism’s capability to produce stem cells may be of importance. In such cases partial growth of new CCPs (bundle branch of the bundle of His or Purkinje fibres) is observed beyond the cicatricial area and results in the regenerated electric impulses conductivity and LVP elimination.
In conclusion I would like to mention that presently the knowledge about the conductive system formation is far from being complete. For example, progress in the study of additional conductive paths for electric impulses was made only due to ECG studies and is not probative. Nobody has ever seen these muscle bundles in a human body! To have a broad picture of all myocardial peculiarities one should know that there is a connective tissue frame between the atriums and the ventricles that prevents the ventricles from excitating together with the atriums. This frame has an innate opening defect of various sizes. It’s this defect that is ablated and not the additional conductivity path (Kent bundle). Pressure increase in an atrium or in a ventricle, or both, makes this defect open and electric impulses pass from atriums to ventricular myocardium from time to time (WPW syndrome). CLC syndrome is also characterized by a lack of additional conductivity path (James bundle). It’s an innate periodic violation of AV delay by electrical impulses node with their accelerated conductivity. In such a case the AV node itself is ablated, and then it is partially cicatrized and slows down the conductivity. So, one may observe that additional conductivity paths between atriums and ventricles are absent. If they did exist, electrical impulses would be permanently passing though them, from birth to death, as far as CCP has no valves, while these pre-excitation syndromes may be transitory.
Literature
1. Buziashvili Y.I., Hananaإ،vili E. M., Asymbekova E.U., etc. Relationship between myocardial viability and availability of late potentials of fibrillation in patients undergoing myocardial infarction. //Cardiology. -2002. - â„– 8. - P. 4-7.
2. Gilyarov M.Y., Sulimov V.A. Treatment of heart rhythm disorders in patients with insufficiency of blood circulation. //BC Cardiology. -2010. -No. 22 (18). - P. 1298-1301.
3. Grishaev S.l. Electrical instability of the myocardium in patients with coronary heart disease. //Russian medical journal. -2003. - â„– 2. - P. 13-18.
4. Murashko V.V., Strutynskij A.V. Electrocardiography: a training manual. //4-e IZD. -M.: MEDpress, 2000. - p 312.
5. Eagles V. Handbook of electrocardiography. //4-e deleted. Ed.-m.: medical news agency. -2004. - p 528.
6. Osadchyy, K.K. depression, anxiety and ischemic heart disease: what you need to know your cardiologist? //Cardiology. -2009. - â„– 5. - P. 70-77.
7. K. Lyadov, T.V. Pأ¢tkina hronoterapiأ¢ antiaritmiؤچeskaأ¢ Adequate in patients with IBS. //2001 Associate Professor. "Cardiology-21".
8. Bigger J.T., Fleiss J.L., Kjeiger R. et al. The relationships among ventricular arrhythmias, left ventricular dysfunction and mortality in the 2 years after myocardial infarction. // Circulation. - 1984. - v.69. - ذ . 250.
9. Buxton A.E., Kirk M.M., Miehaud G.T. Current approaches to evaluation and management of patients with ventricular arrhythmias. // Med Health R I - 2001 Feb - â„– 84(2). - ذ . 58-62.
10. Elhendy A., Sozzi F.B., van Domburg et al. Relation between exercise-induced ventricular arrhythmias and myocardial perfusion abnormalities in patients with intermediate pretest probability of coronary artery disease. // Eur J Nucl Med. - 2000 Mar - â„– 27(3). - ذ . 327-332.
11. Gardner P.I., Ursel P.C., Fenoglio J.J. et al. Electrophysiological and anatomic basis for fractionated electrograms recorded from healed myocardial infarcts. // Circulation. - 1989. - â„– 72. - ذ . 596-611.
12. Kaasik A., Ristimae T., Soopold U. et al. The relationship between left ventricular mass and ventricular late potentials in patients with first myocardial infarction. // J Coronary Artery Disease. - 2001 Oct. - â„– 4(1). ذ . 60.
13. Myeburg R.J., Spooner P.M. Opportunities for sudden death prevention: directions for new clinical and basic research. // Cardiovascular Res - 2001 May - â„– 50(2). - ذ . 177-185.
14. Nademanee K, Intarachot V., Josephson M. et al. Prognostic significance of silent myocardial ischemia in patients with unstable angina. // J. Amer. Coil. Cardiol. - 1987. - v.10. - ذ .1.
15. Podrid Ph.J., Kowey P.R. Handbook of cardiac arrhythmia. // Baltimore, Williams & Wilkins. - 1996. - 459 p.
New defibrillation mechanism.
Rustam G. Habchabov
Dagestan state medical academy, Russia
1, st. Lenina, Makhachkala city, 367000
PhD (medical)
E-mail: rustam033@gmail.com
Abstract. The reason for development of life-threatening arrhythmias leading to sudden and total death provoked by cardiac diseases is still unclear, even though cardiologic research in this regard is being carried all over the word. A damaging of the connective insulation cover of the conductive heart path accompanied by ectopic nodes oxidation may result in life-threatening arrhythmias. Such reason for it as the cardiac electrical instability hasn’t been examined by anyone before. The connective insulation cover prevents a defibrillating electric discharge from penetrating the myocardium. Simultaneously, nobody took the conductive nervous system into account.
Keywords: electric instability of heart; trembling; fibrillation; defibrillation; nervous system; syndrome WPW-CLC.
Cardiovascular diseases (CVDs) remain main causes of invalidity and death of working-age population all over the world. In Russia CVDs frequency is increasing, while our country is considered to be one of the world leaders in death caused by CVDs. All this makes us search for modern and efficient methods of diagnostics, treatment and prevention of the cardiac electrical instability (CEI).
At a young age patients with rheumatic disease, myocarditis, mitral valvular disease demonstrate flutter and fibrillation, while geriatric and older patients demonstrate it more often if they are diagnosed with ischemic heart disease (IHD), myocardial infraction (MI), chronic cardiac failure, mitral stenosis, hypertensive disease, cardiomyopathy, myocarditis, etc. [4].
The condition specifying lethal arrhythmias is structural cardiac pathology (SCP) turning into an instable substrate as affected by various functional factors. Such structural changes conditioning the development of life-threatening arrhythmias (LTA) may be as follows: express hypertrophy, dilatation, cardiac aneurysm, myocardial necrosis and sclerosis, inflammation accompanied by myocardial tissue edema, etc. According to the data of many researchers, these changes constitute the anatomical substrate with various LTA development mechanisms [9].
Many authors that conducted CEI research often concentrate only on myocardial necrotizing and cicatricial lesions (NCL) in IHD patients. Yet such authors ignore other patients with other CVDs that are exposed to total and sudden cardiac death (SCD), too, for no apparent reason.
It should be mentioned that practically all arrhythmias are easy to be analyzed, except for the reason and the mechanism for flutter and fibrillation development. Moreover, the definition of atrial fibrillation is misleading because of the contradictions contained in electrocardiography (ECG) textbooks.
According to the definition contained in ECG textbooks [4,5], atrial and ventricular flutter is regular rhythmical movement of a potent impulse following one and the same path with simultaneous re-entry mechanism development (the reason and the method of such macro-re-entry mechanism being unclear to the researchers at that moment). Atrial and ventricular fibrillation is irregular excitation and contraction of some groups of muscle fibers, each of them being a sort of ectopic impulse site (such definition in ECG textbooks does not seem to be too correct for students and young scientists). Having read the definition and returning back to the “automatism function†chapter one may read that the automatism function is pertinent to Sinoatrial Node cells (SAN) and the conductive system of heart: atrioventricular AV connection, the connective system of atriums and ventricles, the contractive myocardium being deprived of the automatism function [4,5]. All this testifies to the fact that in the event of atrial fibrillation “some groups of muscle fibers†cannot be generated, even if the myocardium or cardiomyocytes change their properties. The “electric myocardial instability†is non-existent because the myocardium can only conduct electric impulses. Some may object that even though the myocardium is deprived of electrical activity, yet it may have a nonhomogeneity area associated with NCL that may prevent it from conducting electricity. Virtually, the electrical impulses avoid the area easily, even in the event of extensive MI, even if there are no serious problems of the cardiac conduction path (CCP). Atrial fibrillation itself is conditioned by multiple generations of ectopic focuses (EF) of a lesser power located in distal CCP and with the same number of colliding micro-re-entry waves. In fact, the myocardium cannot generate it and the ectopic focuses are not chaotically scattered along the myocardium, as some might believe. All EFs are placed along the CCP, as a string of beads, and have the same connective insulation cover (CIC) starting from the SAN. If this cover is intact, electric impulses cannot leave the myocardium. It’s only Purkinje fibers that lack such cover and it’s due to them that the myocardium is excitated.
First of all one should mention that CCPs are placed in the same manner as electric wires in walls of our houses are. They also have an insulation cover. Yet the wires located in walls are immovable and they almost do not wear out while a working myocardium with SCP causes them to wear out soon resulting in a block and/or damaging of CIC only.
In some cases of express SCP over-distension breaking or tearing of a CIC of one of the main proximal CCP may occur. It activates the nearest potent EF with flutter development. This causes a potent electrical impulse to reach the myocardium along the path of least resistance through such damaging and trigger the macro re-entry mechanism. This is how large F flutter waves are formed (besides, the same mechanism is described for Alzheimer disease when atherosclerosis damages the nerve’s medullary sheath and nerve impulses diffuse into the surrounding tissue. Is CCP’s atherosclerotic damaging also possible?).
One may wonder if the proposed mechanism of flutter development is correct. The fact is that the most potent ectopy source in supraventricular paroxysmal tachycardia and atrial flutter is located in the atrium (atrial fibrillation cannot be accounted for; it is conditioned by multiple ectopy). Yet ECG demonstrates a different picture. Why? It may be explained by the fact that in supraventricular paroxysmal tachycardia an electric impulse has to overcome many obstacles passing through a number of small conductive paths and ectopic nodes within the atriums and it reaches the myocardium as a not-so-potent impulse to spin a macro-re-entry wave. In atrial flutter a potent electric impulse reaches the myocardium prematurely, through a CIC damaging, encountering no obstacles and spins a macro re-entry wave. It’s most probable that the lesser part of electric impulse that passes along the conductive paths and exits Purkinje fibres prevents the greater part of electric impulse that reaches the myocardium prematurely, through a CIC damaging, from entering the conductive paths of atriums, encountering a counter-propagating depolarization wave there. All this forms a macro re-entry wave. Such atrial pre-excitation with the macro re-entry mechanism results in a small difference in the cardiac rate between the supraventricular paroxysmal tachycardia and atrial fibrillation. In other words, most frequently paroxysmal tachycardia damages CIC CCP (the conductive paths of Venkenbach, Bakhman and Torel, and the Bundle of His and branches in the ventricles) and becomes flutter. Yet the ectopy source remains to be the same, so it’s the same type of arrhythmia having different mechanisms of action. For example, I have mentioned the atrial process which is exactly the same as the ventricular process.
What is the cause for potent EF activation with flutter development in the event of CIC damaging? If CICs are intact, CCPs are not subject to oxidation. Purkinje, the famous researcher, described transitional T-cells located between the conductive B-cells (Purkinje cells) and the myocardium. He thought that their main function was to conduct electric impulses but their main function is more likely to consist in forming an antioxidative barrier for the conductive B-cells. Myocardial intercellular fluid shall not get into CCP, so the two conductors having a completely different structure have to have an “adaptor†among them that would prevent them from oxidation. If we eliminate the T-cells, the conductivity of electric impulses between B-cells and the myocardium shall remain the same for some time but Purkinje fibres would be exposed to oxidation. With the course of time oxidation would produce irrevocable consequences. Nature did not provide for transitional T-cells for emergency cases. It results in rapid oxidation of the nearest proximal EF with concomitant flutter development. Further the oxidation processes shall reach other distal EF and flutter will gradually turn into fibrillation.
In many cases flutter does not precede atrial fibrillation. It happens because in fibrillation CIC of mid-distal CCPs are to be damaged most frequently (atrial or ventricular Purkinje fibres and braches), especially in NCL. Multiple T-cells damaging is also possible and results in the oxidation of more than one less potent EFs and micro-re-entry mechanism’s development. Thus one may say that the reason for CEI development is the same for flutter and fibrillation.
Any sudden myocardial movements or lesions may trigger CIC CCP damaging in SCP: all types of tachycardia, extrasystoles, blocks, abrupt blood pressure (BP) increase, myocardial contractility increase (especially if accompanied with pathologically decreased contractility), NCL, etc.
As for the focalization, the most thin and easy-to-be-damaged ventricular segments are the right bundle and the left anterior bundle branch. Then main stem of the left branch, the Bundle of His and, finally, the right anterior bundle branch may be named in the descending order. Yet conductivity may be damaged in any segment or in a few segments simultaneously [5].
Other main factor in arrhythmia development is associated with the affected contractility and excitation, most frequently in SCP patients:
1. Organic or functional weakness of the ectopic node (I, II, III block – by AV block type) that result in weak electric impulses arriving to the distal node and its eventual activation.
2. Over-distension of the conductive path (especially in places where ectopic nodes are located) in express hypertrophies, dilatations, etc. lead to the extension of conductivity which also enhances a delay in electric impulses and activates EF.
3. Myocardial inflammation slows down the conductivity and results in ectopic activity development.
The example may be as follows: a city has been growing for 20-30 years because its suburbs (myocardial hypertrophy, dilatation) were growing, too. If the public utility network is lacking in the new districts, the inhabitants of such districts would complain of the lack of electricity. The same is true for the heart. The central nervous system tries to increase the cardiac rate and the potency of electric impulses to compensate for it. Sometimes it is enough. If not, ectopic arrhythmia tries to compensate for such insufficiency.
In some cases electric cardioversion may stop atrial fluttering and fibrillation at their early stages (paroxysmal, persistent). Powerful SAM stimulation suppressing other generation centers takes place. For some time the oxidative influence on EF becomes not important and minute CIC damaging is regenerated after a while. High doses of antyarrhytmic medicines at early stages of flutter and fibrillation are also capable of suppressing ectopic activity with gradual regeneration of minute CIC damaging and sinus rhythm restoration.
Preventive use of repairers in SCP patients may prevent CIC CCP damaging, such repairers being: potassium orotate, ATP and solcoseryl. It goes without saying that basic treatment of patients with atrial flutter and fibrillation shall remain at the previous level– cardioversion and antyarrhytmic drugs. Though additional alkalizing medicines and repairers, with the exception of aspirin and other acid-containing substances, for the period of flutter and fibrillation treatment may contribute into restoring cardiac electrical stability.
To continue CIC and the conductive system’s issue, let me add a few speculations of my own. Electric defibrillation discharges are considered to influence the heart directly, but it’s an error! In the course of defibrillation human anatomy prevents electric discharges from entering the myocardium and exiting from it because of the external epicardial and endocavitary endocardial cardiac layer that has CIC. Electric discharges are most likely to affect the heart in an indirect manner, through multiple nervous receptors. Electric impulses strive to reach the CNS and then brain activation concerning all its aspects, including the sympathic nervous system (SNS) of the heart, starting from and SAN and b-adrenoreceptors stimulation with catecholamines (adrenaline, noradrenaline) discharge. Nobody thought that external electric currents can also take nervous paths in the course of defibrillation.
Now I’m going to present a theory which would seem incredible to many scientists but it has a right to exist. It’s still unknown where and how electric impulses are formed in the nervous system. Electric impulses seem more likely not to be generated by the nervous system but to be sent to it from all the nervous receptors, starting from the SAN and ending up with ventricular myocardium where the electric impulse comes to a dead end. Further electricity is conducted by the conductive (afferent) nerve fiber to various CNS divisions. One should understand that ECG and electroencephalogram (EEG) recording have nothing in common because they record excitation in different structures of the organism. Such circular relationship of the heart and the nervous system is more reasonable that two separate electric systems which would have inevitably come in conflict and lead to a short circuit in one and the same organism.
One may wonder why the nervous system needs electricity. Only for instant processing of the received information and for sending it back. Passive work of the nervous system may be performed without electricity, as it happens during heart transplantation when a patient remains on artificial blood circulation for a few hours. EEG records passive work of the brain which should give an impulse for the transplanted heart to start. In some cases it’s not enough and defibrillation is implemented.
Further we are going to analyze literature sources and main views of the authors and researchers with reference to the CEI issue. First of all one should mention that many authors write “electrical instability of myocardium†while in reality it’s more correct to write “cardiac electrical instabilityâ€.
Life-threatening LTA are caused by a combination of reasons predisposing to electrical instability of myocardium: a substrate (structural cardiac disease) modulating the dysfunction of the autonomous nervous system and LTA triggering factors. The morphologic substrate creating post-MI nonhomogeneity of impulse conductivity is a myocardial area adjoining the necrotized tissue formed by intertangled spots of healthy myocardial fibers and the connective tissue. In this place the impulse connective path is prolonged because the spots of connective tissue serve as barriers to the excitation wave and the conductivity is slowed down due to the affected parallel orientation of muscle fibers. Thus myocardial areas with delayed ventricular depolarization may be anatomic and physiological substrate for the re-entry – main mechanism of LTA development [1,3]. The author of this study was close to understand the real situation but the source of LTA development is not the borderline myocardium but CIC CCP necrotic damaging in the area with concomitant development of flutter and/or atrial fibrillation.
Following the results of the work of J.D. Kramer et al., long impulse spin path is not necessary a small diameter of myocardial tissue with its electrophysiological properties altered by acute myocardial ischemia or with a heterogeneous structure resulting from fibrous and necrotizing changes is enough to trigger the re-entry mechanism [11]. Fibers of the conductive system with EF are more hypoxia-resistant and myocardial ischemia does not trigger the re-entry mechanism in them, while fibrous-necrotizing changes are capable of damaging CIC CCP and triggering arrhythmia with the re-entry mechanism.
As a rule, in the course of the conducted study sudden ventricular tachycardia (VT) or ventricular fibrillation (VF) with the maintained ejection fraction was observed in patients with implanted cardioverters defibrillators; if the contractility is decreased before VT or VF attacks, normally gradual increase of ventricular ectopic activity is to be noted [7]. It goes without saying that cardiac failure is an essential arrhythmogenic factor and a risk marker of sudden arrhythmic death in IHD patients [12]. A Cardiac aneurysm, post-infarction cicatricial changes and clinical manifestations of cardiac failure make the adverse outcome more probable. Left ventricular contractility decrease increases SCD risk not only in IHD but also in patients with other cardiac diseases [8, 9]. Ejection fraction less than 40%, nonsustained ventricular tachycardia (VT) diagnosed by Holter monitoring and electrophysiological study in patients that had an acute MI in their medical history are remaining to be the main predictive markers of high SCD risk [10]. Such combination of two SCD risk factors as frequent ventricular arrhythmia and left ventricular dysfunction with an ejection fraction decrease <40% is especially unfavorable. According to the data of GISSI-2 research, a risk of sudden arrhythmic death in this case increases 16-fold [2, 15].
Besides that factors that were mentioned above, other sudden death risk factors are known, autonomic imbalance of the heart with prevailing sympathetic activity, in particular. The most important markers of the state is a decrease of the cardiac rate variability (CRV) and also such factors as a continued prolongation of the QT interval dispersion and late ventricular potentials (LVP) [13,14].
I can only add a few summarizing conclusion from the PhD thesis I defended with reference to CRV, QT interval dispersion and LVP. Initial degradation of CRV parameters in patients during the post-infarction period is associated with their anxious and depressive state after acute MI. Depression is diagnosed in 82% of patients in the post-infarction period [6]. Such patients are afraid of death and are anxious for their health, they do not perceive their environment with joy. On the contrary, they become reserved and their SNS becomes more active than the parasympathetic nervous system (PNS). As positive psycho-emotional state of the patients is activated, they start to overcome depression and PNS functioning becomes stable, too. This results in the sinus rhythm variations. One should note that a positive psycho-emotional state of a post-MI patient may become negative and even stress condition. This, in turn, leads to SNS hyperarousal. Combined with atherosclerotic changes in coronary arteries, SNS hyperarousal may result in a spasm of coronary arteries and appearance of new necrotic lesions in the myocardium. This is why SAN nervous regulation is more perfect due to the simultaneous operations of the sympathetic and the parasympathetic divisions of the autonomous nervous system.
LVP in the post-infarction period is improved less significantly than the QT interval dispersion parameters. This may be conditioned by the fact that LVP is more associated with the CCP block (His bundle branches and/or main stems of Purkinje fibres) that are completely blocked by the nonhomogeneous necrotized area and further cicatricial changes of the myocardium which make the electric impulse return and reach the myocardium through other CCPs.
As different from LVP, the regeneration processes of the QT interval are to a lesser extent associated with CCP pathological changes. In the acute MI, the QT interval dispersion is predominantly slowed down by necrosis and a reinfarction myocardial area having a nonhomogenious area which is 20-40% larger than in the cicatricial period. Further it improves re-polarization processes. In single cases of LVP improvement in the post-infarction period rare cases of the organism’s capability to produce stem cells may be of importance. In such cases partial growth of new CCPs (bundle branch of the bundle of His or Purkinje fibres) is observed beyond the cicatricial area and results in the regenerated electric impulses conductivity and LVP elimination.
In conclusion I would like to mention that presently the knowledge about the conductive system formation is far from being complete. For example, progress in the study of additional conductive paths for electric impulses was made only due to ECG studies and is not probative. Nobody has ever seen these muscle bundles in a human body! To have a broad picture of all myocardial peculiarities one should know that there is a connective tissue frame between the atriums and the ventricles that prevents the ventricles from excitating together with the atriums. This frame has an innate opening defect of various sizes. It’s this defect that is ablated and not the additional conductivity path (Kent bundle). Pressure increase in an atrium or in a ventricle, or both, makes this defect open and electric impulses pass from atriums to ventricular myocardium from time to time (WPW syndrome). CLC syndrome is also characterized by a lack of additional conductivity path (James bundle). It’s an innate periodic violation of AV delay by electrical impulses node with their accelerated conductivity. In such a case the AV node itself is ablated, and then it is partially cicatrized and slows down the conductivity. So, one may observe that additional conductivity paths between atriums and ventricles are absent. If they did exist, electrical impulses would be permanently passing though them, from birth to death, as far as CCP has no valves, while these pre-excitation syndromes may be transitory.
Literature
1. Buziashvili Y.I., Hananaإ،vili E. M., Asymbekova E.U., etc. Relationship between myocardial viability and availability of late potentials of fibrillation in patients undergoing myocardial infarction. //Cardiology. -2002. - â„– 8. - P. 4-7.
2. Gilyarov M.Y., Sulimov V.A. Treatment of heart rhythm disorders in patients with insufficiency of blood circulation. //BC Cardiology. -2010. -No. 22 (18). - P. 1298-1301.
3. Grishaev S.l. Electrical instability of the myocardium in patients with coronary heart disease. //Russian medical journal. -2003. - â„– 2. - P. 13-18.
4. Murashko V.V., Strutynskij A.V. Electrocardiography: a training manual. //4-e IZD. -M.: MEDpress, 2000. - p 312.
5. Eagles V. Handbook of electrocardiography. //4-e deleted. Ed.-m.: medical news agency. -2004. - p 528.
6. Osadchyy, K.K. depression, anxiety and ischemic heart disease: what you need to know your cardiologist? //Cardiology. -2009. - â„– 5. - P. 70-77.
7. K. Lyadov, T.V. Pأ¢tkina hronoterapiأ¢ antiaritmiؤچeskaأ¢ Adequate in patients with IBS. //2001 Associate Professor. "Cardiology-21".
8. Bigger J.T., Fleiss J.L., Kjeiger R. et al. The relationships among ventricular arrhythmias, left ventricular dysfunction and mortality in the 2 years after myocardial infarction. // Circulation. - 1984. - v.69. - ذ . 250.
9. Buxton A.E., Kirk M.M., Miehaud G.T. Current approaches to evaluation and management of patients with ventricular arrhythmias. // Med Health R I - 2001 Feb - â„– 84(2). - ذ . 58-62.
10. Elhendy A., Sozzi F.B., van Domburg et al. Relation between exercise-induced ventricular arrhythmias and myocardial perfusion abnormalities in patients with intermediate pretest probability of coronary artery disease. // Eur J Nucl Med. - 2000 Mar - â„– 27(3). - ذ . 327-332.
11. Gardner P.I., Ursel P.C., Fenoglio J.J. et al. Electrophysiological and anatomic basis for fractionated electrograms recorded from healed myocardial infarcts. // Circulation. - 1989. - â„– 72. - ذ . 596-611.
12. Kaasik A., Ristimae T., Soopold U. et al. The relationship between left ventricular mass and ventricular late potentials in patients with first myocardial infarction. // J Coronary Artery Disease. - 2001 Oct. - â„– 4(1). ذ . 60.
13. Myeburg R.J., Spooner P.M. Opportunities for sudden death prevention: directions for new clinical and basic research. // Cardiovascular Res - 2001 May - â„– 50(2). - ذ . 177-185.
14. Nademanee K, Intarachot V., Josephson M. et al. Prognostic significance of silent myocardial ischemia in patients with unstable angina. // J. Amer. Coil. Cardiol. - 1987. - v.10. - ذ .1.
15. Podrid Ph.J., Kowey P.R. Handbook of cardiac arrhythmia. // Baltimore, Williams & Wilkins. - 1996. - 459 p.
Article By: Rustam
High Blood Pressure, Causes, and Natural Remedies
Managing Heart Disease The Natural Way
How to Treat Angina Pectoris (Chest Pain)
Eating Low-Fat Dairy Food May Reduce Your Risk Of Stroke
A Relief From Acid Reflux Symptoms Can Be Possible With Generic Prilosec
Want To Shoot Out Hypertension Effectively, Generic Toprol Has The Capability
Do The Arthritis Drugs Celebrex and Vioxx Cause Heart Attacks?
Most Popular Tags
Rustam G Habchabov
,rustam g habchabov
, does did vioxx cause AV heart block, cardiac mechanical instability, cardiac electrical instability, causes of electrical instabiliyy in heart,mi electrical instability arrhythmias
,what are the new cardiopulmonary mechanisms
, ,mechanism of deffibrillation on the heart in vf
,cardiac conduction instability
, , , , ,at young age flutter and fibrillation occur in patients with rheumatism myocarditis
, ,Most Read
New Articles
Most Viewed
Most Downloads
 Nose anatomy
Nose anatomy Humerus bone
Humerus bone Eye anatomy
Eye anatomy Coronary arteries anatomy
Coronary arteries anatomy Female pelvic anatomy
Female pelvic anatomy Heart and lung anatomy
Heart and lung anatomy Bones and ligaments of the FEMALE Pelvis
Bones and ligaments of the FEMALE Pelvis Neck Anatomy
Neck Anatomy MidBrain anatomy
MidBrain anatomy Oral Cavity
Oral Cavity Stomach anatomy
Stomach anatomy Lung anatomy
Lung anatomy Basal Cell Carcinoma ("Rodent Ulcer" Type)
Basal Cell Carcinoma ("Rodent Ulcer" Type)
 Basal Cell Carcinoma ("Rodent Ulcer" Type)
Basal Cell Carcinoma ("Rodent Ulcer" Type) Basal Cell Carcinoma (Histology-Morpheaform Type)
Basal Cell Carcinoma (Histology-Morpheaform Type)
 Basal Cell Carcinoma (Histology-Morpheaform Type)
Basal Cell Carcinoma (Histology-Morpheaform Type) Basal Cell Carcinoma (Histology-Nodular Type - High power)
Basal Cell Carcinoma (Histology-Nodular Type - High power)
 Basal Cell Carcinoma (Histology-Nodular Type - High power)
Basal Cell Carcinoma (Histology-Nodular Type - High power) Basal Cell Carcinoma (Histology-Nodular Type- High power)
Basal Cell Carcinoma (Histology-Nodular Type- High power)
 Basal Cell Carcinoma (Histology-Nodular Type- High power)
Basal Cell Carcinoma (Histology-Nodular Type- High power)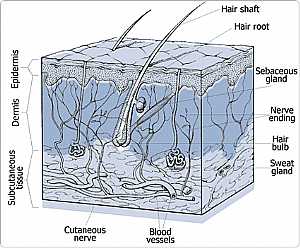 Skin
Skin
 Skin
Skin Nervous System -- Basic
Nervous System -- Basic
 Nervous System -- Basic
Nervous System -- Basic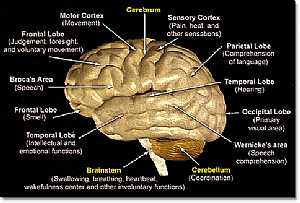 Brain anatomy
Brain anatomy
 Brain anatomy
Brain anatomy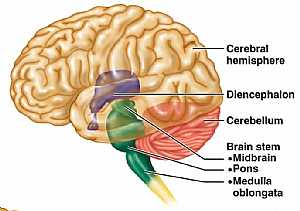 Brain anatomy
Brain anatomy
 Brain anatomy
Brain anatomy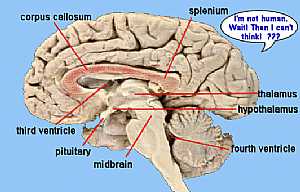 Brain anatomy
Brain anatomy
 Brain anatomy
Brain anatomy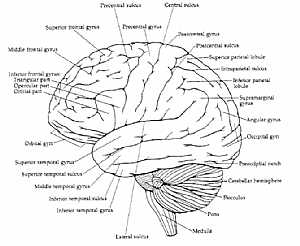 Brain anatomy
Brain anatomy
 Brain anatomy
Brain anatomy Head anatomy
Head anatomy
 Head anatomy
Head anatomy Brain anatomy
Brain anatomy
 Brain anatomy
Brain anatomyeDoctorOnline.com does not provide medical advice, diagnosis or treatment.
© Copyright 2001-2022 eDoctorOnline.com
© Copyright 2001-2022 eDoctorOnline.com
Very nice article. Beautifully present cardiac electrical instability. Keep it up